Drew DeHaas
Thoughts on implementation methods for scalable computational biology software. Updates about Genotype Representation Graphs (GRGs).
GRGL v2.0: improved speed and reduced size
by Drew DeHaas
I recently released the Genotype Representation Graph Library (GRGL) version 2.0. This library is used to create and process graphs that represent the genotype matrix for a dataset, representing the same genotype data that a VCF or BGEN file might. Most representations of genetic datasets are not sparse, but explicitly represent the entire genotype matrix (the mapping between samples and which genetic variants they contain). Compressive genomics formats like GRG outperform even sparse representations (such as IGD), which themselves can already be significantly smaller and faster than non-sparse formats.
The first version of GRGL was described in this paper, and already produced a very small graph file that could be built cheaply at biobank scale. GRG v1.0 was a bit larger than related techniques XSI and Savvy, but more than 3x faster for traversals over the data. This new version of GRG (v2.0) has significant improvements to both size and speed, and is now about the same size as XSI, while often being more than 10x faster. GRG also directly supports performing GWAS calculations, in the same way that other tools do (like plink), and also supports phenotype simulation for testing GWAS calculations/techniques.
One of the benchmarks we use for GRG is a stdpopsim-generated dataset (via msprime) that uses the “OutOfAfrica_4J17” model, and then samples only from the “CEU” population, in our attempt to mimic the UK Biobank dataset. This dataset contains 500,000 diploid samples (1 million haplotypes).
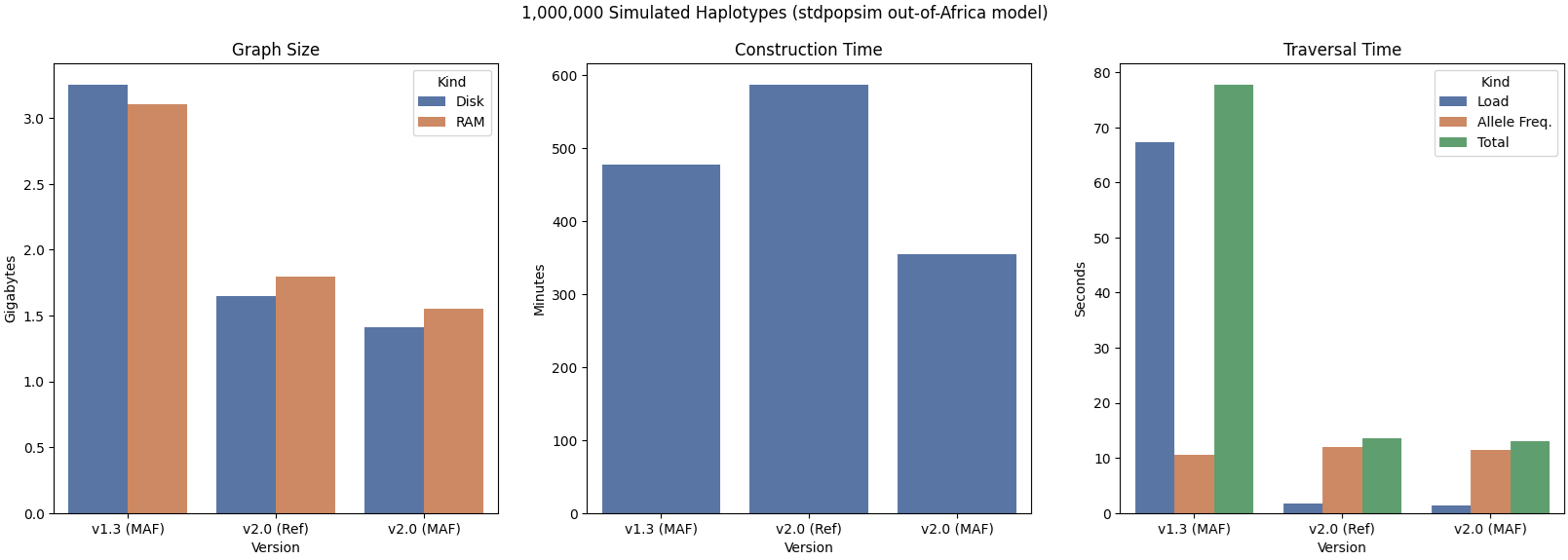
As you can see, both RAM and disk usage are halved with v2.0. Construction time is also faster in an apples-to-apples comparison. The “Ref” and “MAF” versions shown in the above graph reflect whether the GRG was built with the default options or with --maf-flip. The default options will maintain the reference allele for all variants in the dataset, in the GRG. This can be useful if your downstream processing needs a particular reference (e.g. GRCh38), or more realistically if you have polarized data and want your GRG Mutation nodes to truly represent mutations that have occurred in more recent time. In contrast, --maf-flip will just create mutation nodes such that the reference is always the most frequent allele. As you can imagine, from a data compression perspective, this makes the GRG construction problem easier.
See the GRGL v2.0 release notes for more information about what’s changed in v2.0. If you’re interested in learning more about GRG, and what you can do with it, check out the documentation and learn how you can construct GRGs, convert the excellent tskit format into a GRG, or run GWAS.
tags: GRG - "software - release"